
BRAFTOVI 75 mg, gélule, boîte de 7 plaquettes de 6
Retiré du marché le : 01/12/2020
Dernière révision : 09/07/2020
Taux de TVA : 0%
Laboratoire exploitant : PIERRE FABRE MEDICAMENT
Source :

L'encorafenib est indiqué en association au cetuximab, dans le traitement de patients adultes atteints de cancer colorectal (CCR) métastatique porteur d'une mutation BRAF V600E, ayant progressé après un ou deux traitement(s) antérieur(s) systémique(s) au stade métastatique et présentant un score ECOG 0 ou 1.
Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Composition. Pour les contre-indications au cetuximab, se référer au RCP du cetuximab.
Pour les mises en garde spéciales et précautions liées à l'utilisation du cetuximab, se référer au RCP du cetuximab.
L'encorafenib doit être utilisé en association au cetuximab (chez les patients atteints d'un cancer colorectal métastatique porteur de la mutation BRAF V600E). Pour davantage d'informations sur les mises en garde et précautions associées au traitement par le cetuximab, voir la rubrique Mises en garde et précautions d'emploi du RCP du cetuximab.
Détermination du statut mutationnel BRAF
Avant toute administration de l'encorafenib, les patients doivent être atteints de cancer colorectal métastatique porteur de la mutation BRAF V600E confirmé par un test validé. L'efficacité et la sécurité de l'encorafenib ont été établies uniquement chez les patients porteurs de cancer colorectal exprimant la mutation BRAF V600E. L'encorafenib ne doit pas être utilisé chez les patients atteints d'un cancer colorectal non porteur de la mutation BRAFV600E.
Hémorragie
Des hémorragies, y compris des accidents hémorragiques majeurs, peuvent survenir lors de l'administration d'encorafenib (voir rubrique Effets indésirables). Le risque hémorragique peut augmenter en cas d'utilisation concomitante d'un traitement anticoagulant et antiplaquettaire. La survenue d'accidents hémorragiques de grade ≥ 3 doit être prise en charge avec l'interruption de la dose ou en l'arrêt définitif du traitement (voir Tableau 3 à la rubrique Posologie et mode d'administration) et selon le tableau clinique.
Toxicités oculaires
Des toxicités oculaires, incluant des uvéites, des iritis et des iridocyclites, peuvent se produire lors de l'administration d'encorafenib.
À chaque visite, les symptômes visuels doivent être évalués chez les patients. Si des symptômes indiquant l'apparition ou l'aggravation de troubles de la vision, notamment une baisse de la vision centrale, une vision trouble ou une perte de la vue, sont identifiés, il est recommandé de procéder rapidement à un examen ophtalmologique.
En cas de survenue d'une uvéite, y compris d'une iridocyclite et d'une iritis pendant le traitement, voir la rubrique Posologie et mode d'administration.
Allongement de l'intervalle QT
Un allongement de l'intervalle QT a été observé chez les patients traités par des inhibiteurs de BRAF. Il n'a pas été conduit d'étude dédiée de l'intervalle QT visant à évaluer le potentiel d'allongement de l'intervalle QT par l'encorafenib.
Globalement, les résultats suggèrent que l'encorafenib administré seul est susceptible de provoquer de légères augmentations de la fréquence cardiaque. Les résultats des études groupées de l'encorafenib en association au binimetinib administrés aux doses recommandées et d'une étude d'encorafenib seul, suggèrent que l'encorafenib peut entraîner de légers allongements de l'intervalle QTc (voir rubrique Propriétés pharmacodynamiques).
Les données sont insuffisantes pour exclure un allongement de l'intervalle QT cliniquement significatif dépendant de l'exposition systémique.
En raison du risque potentiel d'allongement de l'intervalle QT, il est recommandé de :
Corriger les anomalies électrolytiques sériques, notamment celles du magnésium et du potassium, de contrôler les facteurs de risque d'allongement de l'intervalle QT (par ex., insuffisance cardiaque congestive, bradycardie) avant le début du traitement et pendant le traitement, et prévoir une surveillance étroite, notamment électrocardiographique étroite en cas de facteurs de risque associés de ce type, incluant les troubles digestifs tels que les diarrhées et ou vomissement susceptibles pouvant fréquemment être à l'origine de trouble hydro-électrolytiques,
Concernant les médicaments allongeant le QTc, d'éviter leur usage associé. Lorsque de tels médicaments doivent être maintenus, ou ne peuvent être substitués par des médicaments dénués d'un tel effet, une surveillance électrocardiographique étroite devra être mise en place,
Il est recommandé de réaliser un électrocardiogramme (ECG) avant le début du traitement par l'encorafenib, un mois après, puis tous les mois ou plus fréquemment au cours du traitement si cliniquement indiqué. Cette surveillance éléctrocardiographique devra être renforcée en cas de facteurs de risques associés (troubles hydroélectrolytiques, médicaments allongeant le QTc, médicament bradycardisant favorisant le potentiel d'allongement du QTc, etc.). La survenue d'un allongement de l'intervalle QTc peut être prise en charge avec une réduction de dose, une suspension de dose ou l'arrêt définitif du traitement ainsi qu'une correction des anomalies électrolytiques et un contrôle des facteurs de risque et/ou une modification des médicaments concomitants potentialisant ce risque (voir rubrique Posologie et mode d'administration). Le recours à un avis spécialisé d'un médecin compétent en trouble du rythme et de la condition est recommandé en cas de survenue d'un allongement du QTc pour discuter le maintien sous traitement, la réduction de dose ou arrêt, ainsi que l'exploration des facteurs favorisant ce risque.
Nouvelles tumeurs primitives
De nouvelles tumeurs primitives, cutanées et non cutanées, ont été observées chez des patients traités par des inhibiteurs de BRAF et peuvent apparaître lors de l'administration de l'encorafenib (voir rubrique Effets indésirables).
Tumeurs cutanées
Des tumeurs cutanées, telles qu'un carcinome épidermoïde cutané (CEC), incluant des cas de kératoacanthome, ont été observées chez des patients traités par des inhibiteurs de BRAF, dont l'encorafenib.
De nouveaux mélanomes primitifs ont été observés chez des patients traités par des inhibiteurs de BRAF, dont l'encorafenib (voir rubrique Effets indésirables).
Un examen clinique dermatologique doit être effectué avant le début du traitement par encorafenib, puis tous les mois pendant le traitement et jusqu'à 6 mois après l'arrêt de celui-ci avec consultation d'un spécialiste en cas de doute. Un examen cutané par un médecin compétent en dermatologie est recommandé après 3 mois de traitement. Les lésions suspectes de la peau doivent être traitées par exérèse cutanée avec un examen dermato-anatomopathologique. Les patients doivent être avertis qu'ils doivent immédiatement signaler à leurs médecins l'apparition de toute nouvelle lésion cutanée. Le traitement par l'encorafenib doit être poursuivi sans modifications de dose.
Tumeurs non cutanées
En raison de son mécanisme d'action, l'encorafenib peut favoriser l'apparition de tumeurs associées à l'activation du gène RAS via des mutations ou d'autres mécanismes. Les patients recevant l'encorafenib doivent bénéficier d'un examen clinique de la tête et du cou, d'un scanner thoraco-abdominal et d'un examen de la région anale et pelvienne (pour les femmes), ainsi que d'une numération de la formule sanguine complète avant le début, au cours et à la fin du traitement, comme cliniquement approprié. L'arrêt définitif de l'encorafenib doit être envisagé chez les patients qui développent des tumeurs non cutanées avec mutation du gène RAS. Les bénéfices et risques doivent être évalués attentivement avant d'administrer l'encorafenib aux patients ayant des antécédents de cancer ou atteints d'un cancer évolutif associé à une mutation du gène RAS.
Anomalies biologiques hépatiques
Les anomalies biologiques hépatiques, notamment une élévation des transaminases ASAT et ALAT ont été observées avec l'encorafenib (voir rubrique Effets indésirables). Les paramètres biologiques hépatiques doivent faire l'objet d'un contrôle avant le début du traitement par encorafenib et faire l'objet d'une surveillance au moins mensuelle pendant le traitement, et plus souvent si cliniquement indiqué. Les anomalies biologiques hépatiques doivent être prises en charge avec une réduction de dose, une suspension de dose ou l'arrêt définitif du traitement (voir rubrique Posologie et mode d'administration).
Insuffisance hépatique
Compte tenu du métabolisme principal de l'encorafenib et de son élimination par le foie, les patients atteints d'une insuffisance hépatique légère à sévère peuvent avoir une exposition systémique à l'encorafenib au-delà de la marge d'exposition liée à la variabilité interindividuelle (voir rubrique Propriétés pharmacocinétiques).
En l'absence de données cliniques, l'encorafenib n'est pas recommandé chez les patients atteints d'une insuffisance hépatique modérée ou sévère.
L'administration d'encorafenib doit être effectuée avec précaution à une dose de 300 mg une fois par jour chez les patients atteints d'une insuffisance hépatique légère (voir rubrique Posologie et mode d'administration).
Une surveillance plus étroite des toxicités liées à l'encorafenib chez les patients atteints d'une insuffisance hépatique légère est recommandée, notamment un examen clinique et un bilan hépatique avec évaluations de l'ECG doit se faire tous les mois pendant le traitement.
Insuffisance rénale
Aucune donnée clinique n'est disponible chez les patients présentant une insuffisance rénale sévère (voir rubriques Posologie et mode d'administration et Propriétés pharmacocinétiques).
L'encorafenib doit être utilisé avec précaution chez les patients présentant une insuffisance rénale sévère. Une augmentation de la créatinine a été fréquemment rapportée avec l'encorafenib administré seul ou en association au cetuximab. Les cas observés de défaillance rénale, y compris d'atteinte rénale aiguë et d'insuffisance rénale, étaient généralement associés à des vomissements et à une déshydratation. D'autres facteurs contributifs, incluent diabète et hypertension artérielle. La créatinine sanguine doit être surveillée tous les mois et l'élévation de la créatinine doit être prise en charge avec une réduction de dose, une suspension de dose ou l'arrêt définitif du traitement (voir Tableau 3 à la rubrique Posologie et mode d'administration). Les patients doivent veiller à avoir une bonne hydratation pendant le traitement.
Effets d'autres médicaments sur l'encorafenib
L'utilisation concomitante d'inhibiteurs puissants du CYP3A pendant le traitement par encorafenib doit être évitée. Si l'utilisation concomitante d'un inhibiteur puissant du CYP3A est nécessaire, il convient de surveiller attentivement les patients pour leur sécurité (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions).
L'administration concomitante d'un inhibiteur modéré du CYP3A et de l'encorafenib doit faire l'objet d'une utilisation avec prudence.
Résumé du profil de sécurité
Le profil de sécurité de l'encorafenib en monothérapie (300 mg oralement une fois par jour) repose sur les données provenant de 217 patients atteints d'un mélanome métastatique ou non résécable porteur d'une mutation BRAF V600 (ci-après désignées comme l'ensemble de la population Enco 300). Les effets indésirables les plus fréquents (> 25 %) rapportés avec l'encorafenib 300 étaient les suivants : hyperkératose, alopécie, érythrodysesthésie palmo-plantaire, fatigue, rash, arthralgies, sécheresse cutanée, nausées, myalgies, céphalées, vomissements et prurit.
La sécurité d'emploi de l'encorafenib (300 mg pris oralement une fois par jour) en association au cetuximab (dosé conformément à son RCP) a été évaluée chez 216 patients atteints de cancer colorectal métastatique porteur d'une mutation BRAF V600E, sur la base de l'étude de phase III ARRAY-818-302. Les effets indésirables les plus fréquents (> 25 %) rapportés dans cette population étaient les suivants : fatigue, nausées, diarrhée, dermatite acnéiforme, douleur abdominale, arthralgies/douleur musculosquelettique, baisse de l'appétit et rash.
Le taux d'arrêt de tous les médicaments de l'étude dû à un effet indésirable était de 1,4 % chez les patients traités par encorafenib en association au cetuximab.
Liste des effets indésirables sous forme de tableau
Les effets indésirables sont énumérés ci-dessous selon les classes de systèmes d'organes MedDRA et les conventions de fréquence suivantes : très fréquent (³1/10), fréquent (³ 1/100 à < 1/10), peu fréquent(³ 1/1 000 à < 1/100), rare (³ 1/10 000 à < 1/1 000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles).
Pour chaque catégorie de fréquence, les effets indésirables sont présentés en ordre décroissant de sévérité.
Tableau 4 : Effets indésirables
| Fréquence | Encorafenib en monothérapie 300 mg (n = 217)** | Encorafenib 300 mg en association au cetuximab (n = 216) |
| Tumeurs bénignes, malignes et non précisées | ||
| Très fréquent | Papillome cutané* Nævus mélanocytaire | Nævus mélanocytaire |
| Fréquent | CEC a Nouveau mélanome primitif* | CECa Papillome cutané* Nouveau mélanome primitif* |
| Peu fréquent | Carcinome basocellulaire | Carcinome basocellulaire |
| Affections du système immunitaire | ||
| Fréquent | Hypersensibilitéb | Hypersensibilitéb |
| Troubles du métabolisme et de la nutrition | ||
| Très fréquent | Baisse de l'appétit | Baisse de l'appétit |
| Affections psychiatriques | | |
| Très fréquent | Insomnie | Insomnie |
| Affections du système nerveux | ||
| Très fréquent | Céphalées* Neuropathie périphérique* Dysgueusie* | Neuropathie périphérique* Céphalées* |
| Fréquent | Parésie faciale | Sensations vertigineuses* Dysgueusie* |
| Affections oculaires | ||
| Peu fréquent | Uvéite* | |
| Affections cardiaques | ||
| Fréquent | Tachycardie supraventriculairec | Tachycardie supraventriculairec |
| Affections vasculaires | ||
| Très fréquent | | Hémorragieg |
| Fréquent | | |
| Affections gastro-intestinales | ||
| Très fréquent | Nausées Vomissements* Constipation | Nausées Vomissements* Constipation Douleur abdominale* Diarrhée* |
| Peu fréquent | Pancréatite* | Pancréatite* |
| Affections de la peau et du tissu sous-cutané | ||
| Très fréquent | Érythrodysesthésie palmo-plantaire (EPP) Hyperkératose* Rash* Sécheresse cutanée* Prurit* Alopécie* Érythèmed Hyperpigmentation cutanée* | Dermatite acnéiforme* Rash* Sécheresse cutanée* Prurit* |
| Fréquent | Dermatite acnéiforme* Exfoliation cutanéee Photosensibilité* | Hyperpigmentation cutanée* Érythrodysesthésie palmo-plantaire (EPP) Hyperkératose* Alopécie* Érythèmed |
| Peu fréquent | | Exfoliation cutanéee |
| Affections musculo-squelettiques et systémiques | ||
| Très fréquent | Arthralgies* Myalgief Douleurs aux extrémités Dorsalgie | Arthralgies*/Douleurs musculosquelettiques* Myopathie/Troubles musculaires* Douleurs aux extrémités Dorsalgie |
| Fréquent | Arthrite* | |
| Affections du rein et des voies urinaires | ||
| Fréquent | Insuffisance rénale* | Insuffisance rénale* |
| Troubles généraux et anomalies au site d'administration | ||
| Très fréquent | Fatigue* Fièvre* | Fatigue* Fièvre* |
| Investigations | ||
| Très fréquent | Gamma-glutamyltransférase (GGT) augmentée* | |
| Fréquent | Transaminase augmentée* Créatinine sanguine augmentée* Lipase augmentée | Créatinine sanguine augmentée* Transaminase augmentée* |
| Peu fréquent | Amylase augmentée | Amylase augmentée Lipase augmentée |
* Noms composés qui comprenaient plusieurs Termes Préférés
** Concerne encorafenib en monothérapie (300 mg oralement une fois par jour) sur la base des données provenant de 217 patients atteints d'un mélanome métastatique ou non résécable porteur d'une mutation BRAF V600
a Comprend, sans s'y limiter, kératoacanthome, carcinome épidermoïde
b Comprend, sans s'y limiter, oedème de Quincke, hypersensibilité médicamenteuse, hypersensibilité, vascularite allergique, urticaire et réaction anaphylactique
c Comprend, sans s'y limiter, extrasystoles, tachycardie sinusale,
d Comprend érythème, érythème généralisé, érythème plantaire
e Comprend dermatite exfoliatrice, exfoliation cutanée, rash avec exfoliation
f Comprend myalgies, fatigue musculaire, lésions musculaires, spasme musculaire, fatigue musculaire
g Comprend hémorragie à différents sites y compris hémorragie cérébrale
Description de certains effets indésirables
Tumeurs cutanées
Carcinome épidermoïde cutané (CEC)
Chez les patients traités par encorafenib en association au cetuximab, des CEC, y compris les kératoacanthomes, ont été observés chez 1,4 % des patients (3/216). Les délais de survenue du premier événement de CEC (tous grades confondus) étaient de 0,5, 0,6 et 3,6 mois pour ces 3 patients.
Nouveau mélanome primitif
Chez les patients traités par encorafenib en association au cetuximab, des événements de nouveaux mélanomes primitifs se sont produits chez 1,9 % des patients (4/216) et ont été déclarés comme étant de grade 2 chez 0,9 % des patients (2/216) et de grade 3 chez 0,9 % des patients (2/216).
Atteintes oculaires
Dans l'ensemble de la population Enco 300, une uvéite a été rapportée chez 0,5% des patients (1/274), de grade 2. Des troubles visuels, y compris une vision trouble et une baisse de l'acuité visuelle sont survenus chez 5,5 % des patients (12/217).
Hémorragie
Des événements hémorragiques ont été observés chez 21,3 % des patients (46/216) traités par encorafenib en association au cetuximab ; 1,4 % des événements (3/216) étaient de grade 3 et un cas fatal a été rapporté. Des interruptions ou des réductions de dose ont été nécessaires chez 1,9 % des patients (4/216). Les événements hémorragiques ont conduit à l'arrêt du traitement chez 1 patient (0,5 %).
Les événements hémorragiques les plus fréquents étaient une épistaxis chez 6,9 % des patients (15/216), une hématochézie chez 2,8% des patients (6/216) et une rectorragie chez 2,8 % des patients (6/216).
Pancréatite
Dans la population traitée par encorafenib en association au cetuximab, une pancréatite de grade 3 avec événements de lipase et amylase augmentées a été rapportée chez 1 patient (0,5 %) et a conduit à l'interruption de la dose.
Réactions cutanées
Rash
Chez les patients traités par encorafenib en association au cetuximab, un rash est survenu chez 30,6 % des patients (66/216). La plupart des événements étaient sans sévérité, un événement de grade 3 ayant été rapporté chez 0,5 % des patients (1/216). Le rash a conduit à une interruption de la dose chez 0,5 % des patients (1/216).
Érythrodysesthésie palmo-plantaire
Dans la population traitée par encorafenib en association au cetuximab, une érythrodysesthésie palmo- plantaire a été rapportée chez 5,1 % des patients (11/216). La plupart des effets de type érythrodysesthésie palmo-plantaire étaient de grade 1 chez 3,7 % des patients (8/216). Des événements de grade 2 ont été rapportés chez 0,9 % des patients (2/216) et de grade 3 chez 0,5% des patients (1/216). Aucune interruption de la dose, modification de la dose ni aucun arrêt définitif du traitement n'a été nécessaire.
Dermatite acnéiforme
Chez les patients traités par encorafenib en association au cetuximab, une dermatite acnéiforme a été rapportée chez 33,3 % des patients (72/216) et était, dans la plupart des cas, de grade 1 (25,5 % des patients (55/216)), ou de grade 2 (6,9 % des patients (15/216)). Une réduction ou une interruption de la dose a été rapportée chez 2,3 % des patients (5/216). Aucun arrêt définitif du traitement n'a été rapporté. La dermatite acnéiforme était généralement réversible.
Photosensibilité
Dans l'ensemble de la population Enco 300, la photosensibilité a été observée chez 4,1 % des patients (9/217). La plupart des événements étaient de grade 1 à 2. Aucun événement n'a requis d'arrêt définitif, de modification ou d'interruption de la dose.
Parésie faciale
Dans l'ensemble de la population Enco 300 traitée pour un mélanome, la parésie faciale a été observée chez 7,4 % des patients (16/217). La plupart des événements étaient légers à modérés : de grade 1 chez 2,3 % des patients (5/217), de grade 2 chez 3,7 % des patients (8/217) et de grade 3 chez 1,4 % des patients (3/217). Le délai médian de survenue de la parésie faciale était de 0,3 mois (de 0,1 à 12,1 mois). La parésie faciale était généralement réversible et a mené à un arrêt définitif du traitement chez 0,9 % des patients (2/217). Une interruption ou une modification de la dose a été observée chez 3,7 % des patients (8/217) et le traitement symptomatique dont les corticostéroïdes a été observé chez 5,1 % des patients (11/217).
Insuffisance rénale
Une augmentation de la créatinine sanguine a été rapportée chez 2,8 % des patients (6/216) traités par encorafenib en association au cetuximab. Tous les événements étaient sans sévérité, à l'exception d'un événement de grade 4. Des événements à type d'insuffisance rénale étaient de grade 3 ou 4 et ont été rapportés comme une atteinte rénale aiguë chez 1,9 % des patients (4/216) et comme une insuffisance rénale chez 0,5 % des patients (1/216).
Anomalies biologiques hépatiques
L'incidence de l'augmentation des transaminases chez les patients traités par encorafenib en association au cetuximab était de 8,8 % des patients (19/216), avec des événements de grade 3 chez 1,4 % des patients (3/216).
Affections gastro-intestinales
Chez les patients traités par encorafenib en association au cetuximab, une diarrhée a été rapportée chez 38,4 % des patients (83/216) et était de grade 3chez 2,8 % des patients (6/216). La diarrhée a conduit à l'arrêt définitif du traitement chez 0,5 % des patients (1/216) et à une interruption ou une modification de la dose chez 3,7 % des patients (8/216).
Une douleur abdominale a été rapportée chez 36,6 % des patients (79/216) et était de grade 3 chez 5,1 % des patients (11/216). Des nausées sont survenues chez 38 % des patients (82/216) avec un grade 3 observé chez 0,5 % des patients (1/216). Des vomissements sont survenus chez 27,3 % des patients (59/216) avec un grade 3 rapporté chez 1,4 % des patients (3/216). Une constipation de grade 1 ou 2 est survenue chez 18,1 % des patients (39/216).
Les affections gastro-intestinales ont généralement été traitées selon les thérapies usuelles.
Céphalée
Chez les patients traités par encorafenib en association au cetuximab, une céphalée s'est produite chez 20,4 % des patients (44/216) et était de grade 1 ou 2.
Fatigue
Chez les patients traités par encorafenib en association au cetuximab, la fatigue a été rapportée chez 56,9 % des patients (123/216), dont 7,9 % (17/216) ont signalé une fatigue de grade 3.
Populations spéciales
Personnes âgées
Parmi les patients traités par encorafenib en association au cetuximab (n = 216), 134 patients (62 %) étaient âgés de moins de 65 ans, 62 patients (28,7 %) étaient âgés de 65 à 74 ans et 20 patients (9,3 %) étaient âgés de 75 ans et plus. Les évènements indésirables les plus fréquents rapportés avec une incidence plus élevée chez les patients âgés de 65 ans ou plus comparés aux patients âgés de moins de 65 ans comprenaient : baisse de l'appétit, anémie, asthénie et dyspnée.
Dans les populations atteintes de cancer colorectal, étant donné le très faible nombre de patients traités dans le sous-groupe d'âge 75 ans et plus, les différences d'incidence des événements indésirables entre ce groupe et le groupe de patients âgés de moins de 75 ans n'ont pu être évaluées.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé doivent déclarer tout effet indésirable suspecté à l'aide de la fiche de déclaration des effets indésirables disponible dans le Protocole d'utilisation thérapeutique et de recueil d'informations (voir Annexe D du PUT)..
CONFIRMER,
avant traitement que le patient est porteur de la mutation BRAF V600 par un
test validé.
SURVEILLANCE du traitement :
- A chaque
visite, contrôler les symptômes visuels du patient.
-
Réaliser un électrocardiogramme avant le début du traitement par l'encorafenib,
un mois après, puis tous les 3 mois environ ou plus fréquemment au cours du
traitement si cliniquement indiqué.
- Faire un examen clinique dermatologique avant le
début du traitement par binimetinib, tous les mois pendant le traitement et
jusqu'à 6 mois maximum après l'arrêt de celui-ci. Les lésions suspectes de la
peau doivent être traitées par exérèse cutanée avec un examen
dermato-anatomopathologique.
- Faire un examen clinique de la tête et du cou, un scanner thoraco-abdominal
et un examen de la région anale et pelvienne (pour les femmes) et une
numération de la formule sanguine complète avant le début, au cours et à la fin
du traitement, comme cliniquement approprié.
- Mesurer
les paramètres biologiques hépatiques avant le début du traitement par
binimetinib, mensuellement pendant le traitement, puis si
cliniquement indiqué.
Femmes en âge de procréer/Contraception chez les femmes
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement par encorafenib et pendant au moins 1 mois après la dernière dose. L'encorafenib peut réduire l'efficacité des contraceptifs hormonaux (voir rubrique Interactions avec d'autres médicaments et autres formes d'interactions). Par conséquent, il est conseillé aux patientes qui ont recours à une contraception hormonale d'utiliser une méthode alternative ou supplémentaire telles qu'une méthode barrière (un préservatif, par exemple) pendant le traitement par encorafenib et pendant au moins 1 mois après la dernière dose.
Grossesse
Il n'existe pas de données sur l'utilisation de l'encorafenib chez la femme enceinte. Les études effectuées chez l'animal ont mis en évidence une toxicité sur la reproduction (voir rubrique Données de sécurité précliniques).
L'encorafenib n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception. Si l'encorafenib est utilisé pendant la grossesse ou si une grossesse survient pendant le traitement, la patiente devra être informée du danger potentiel pour le foetus.
Allaitement
On ne sait pas si l'encorafenib ou ses métabolites sont excrétés dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut être exclu. Une décision doit être prise soit d'interrompre l'allaitement soit d'interrompre le traitement avec encorafenib en prenant en compte le bénéfice de l'allaitement pour l'enfant au regard du bénéfice du traitement pour la mère.
Fertilité
Il n'existe pas de donnée sur les effets de l'encorafenib sur la fertilité chez les humains. Selon les résultats obtenus chez les animaux, l'utilisation d'encorafenib pourrait avoir un effet sur la fertilité des hommes présentant un potentiel reproductif (voir rubrique Données de sécurité précliniques). Comme la pertinence clinique de ces observations n'est pas connue, les patients de sexe masculin doivent être informés du risque éventuel d'une spermatogenèse altérée.
Effets d'autres médicaments sur l'encorafenib L'encorafenib est principalement métabolisé par CYP3A4. Inhibiteurs du CYP3A4
L'administration concomitante d'inhibiteurs du CYP3A4 modérés (diltiazem) et puissants (posaconazole) avec des doses uniques d'encorafenib chez des volontaires sains a entraîné le doublement et le triplement de l'aire sous la courbe de la concentration en fonction du temps (ASC)), respectivement, et une augmentation de 44,6 % et 68,3 % de la concentration maximale d'encorafenib, respectivement. Des prédictions reposant sur les modèles indiquent que l'effet du posaconazole après des administrations répétées pourrait être similaire pour l'ASC (augmentation multipliée par 3) et légèrement plus puissant pour la Cmax (augmentation multipliée par 2,7). Des prédictions reposant sur des modèles pour le kétoconazole suggèrent une augmentation multipliée par environ 5 pour l'ASC de l'encorafenib et une multiplication par 3 à 4 pour la Cmax de l'encorafenib après administration de doses de 450 et 300 mg d'encorafenib une fois par jour, respectivement.
Par conséquent, l'administration concomitante d'encorafenib et d'inhibiteurs puissants du CYP3A4 doit être évitée en raison de l'exposition accrue à l'encorafenib et de l'augmentation possible de la toxicité, (voir rubrique Propriétés pharmacocinétiques). Exemples non exhaustifs d'inhibiteurs puissants du CYP3A4 : le ritonavir, l'itraconazole, la clarithromycine, la télithromycine, le posaconazole et le jus de pamplemousse. Si l'usage concomitant d'un inhibiteur puissant du CYP3A est inévitable, il convient de surveiller attentivement les patients pour garantir leur sécurité.
L'administration concomitante d'inhibiteurs modérés du CYP3A4 devra être effectuée avec précaution. Exemples non exhaustifs d'inhibiteurs modérés du CYP3A4 : l'amiodarone, l'érythromycine, le fluconazole, le diltiazem, l'amprénavir et l'imatinib. Lors de l'administration concomitante d'encorafenib et d'un inhibiteur modéré du CYP3A4, il convient de surveiller attentivement les patients pour garantir leur sécurité.
Inducteurs du CYP3A4
L'administration concomitante d'encorafenib et d'un inducteur du CYP3A4 n'a pas été évaluée dans une étude clinique. Cependant, une réduction de l'exposition à l'encorafenib est probable et risquerait d'en compromettre l'efficacité. Voici une liste non exhaustive d'exemples d'inducteurs modérés ou puissants du CYP3A4 : la carbamazépine, la rifampicine, la phénytoïne et le millepertuis. D'autres agents ne présentant aucune induction ou une induction minimale du CYP3A doivent être envisagés.
Effets de l'encorafenib sur d'autres médicaments
Substrats du CYP
L'encorafenib est à la fois un inhibiteur et un inducteur du CYP3A4. L'usage concomitant avec des agents qui sont des substrats du CYP3A4 (par ex., des contraceptifs hormonaux) risque d'entraîner une hausse de la toxicité ou une perte de l'efficacité de ces agents. L'administration concomitante d'agents qui sont des substrats du CYP3A4 devra être effectuée avec précaution.
L'encorafenib est un inhibiteur de l'UGT1A1. Les agents concomitants qui sont des substrats de l'UGT1A1 (tels que le raltégravir, l'atorvastatine, le dolutégravir) risquent de présenter une exposition accrue et doivent donc être administrés avec précaution.
Substrats de transporteurs
L'encorafenib a la capacité d'inhiber un certain nombre de transporteurs. Les agents qui sont des substrats des transporteurs rénaux OAT1, OAT3, OCT2 (tels que le furosémide, la pénicilline) ou des agents qui sont des substrats des transporteurs hépatiques OATP1B1, OATP1B3, OCT1 (tels que l'atorvastatine, le bosentan) ou des substrats de la BCRP (tels que le méthotrexate, la rosuvastatine) ou des substrats de la P-gp (tels que le pozaconazole) risquent de présenter une exposition accrue et leur administration concomitante doit donc être effectuée avec précaution.
Le traitement par l'encorafenib doit être initié et supervisé par un médecin expérimenté dans l'utilisation de médicaments anticancéreux.
Posologie
La dose recommandée d'encorafenib est de 300 mg (quatre gélules de 75 mg) une fois par jour, lorsqu'il est associé au cetuximab.
Adaptation posologique
La prise en charge des effets indésirables peut nécessiter une réduction de dose, une interruption temporaire ou un arrêt définitif du traitement par encorafenib (voir Tableaux 1 et 2 et 3 ci-dessous).
Pour toute information sur la posologie et les recommandations de réduction de dose pour le cetuximab, voir rubrique Posologie et mode d'administration du RCP du cetuximab.
Les recommandations de réduction de dose pour l'encorafenib sont présentées dans le Tableau 1.
Tableau 1: Modifications de dose recommandées pour l'encorafenib quand utilisé en association au cetuximab dans l'indication du CCR
| Niveau de dose | Dose d'encorafenib utilisé en association au cetuximab |
| Dose initiale | 300 mg une fois par jour |
| 1ère réduction de dose | 225 mg une fois par jour |
| 2nde réduction de dose | 150 mg une fois par jour |
Si l'encorafenib est définitivement arrêté, le cetuximab doit également être définitivement arrêté. Si le cetuximab est définitivement arrêté, l'encorafenib doit également être définitivement arrêté.
Les adaptations posologiques en cas d'effets indésirables sont indiquées dans les tableaux 2 et 3 ci- dessous.
En cas de survenue de nouvelles tumeurs cutanées primitives: aucune modification de dose n'est requise pour l'encorafenib.
En cas de survenue de nouvelles tumeurs non cutanées primitives positives à la mutation RAS : l'arrêt définitif du traitement par encorafenib doit être envisagé.
Tableau 2 : Adaptations posologiques recommandées pour l'encorafenib (utilisé en association au cetuximab) pour certains effets indésirables
| Sévérité des effets indésirablesa | Encorafenib |
| Réactions cutanées | |
| Grade 2 | L'encorafenib doit être maintenu. Si l'éruption cutanée s'aggrave ou ne s'améliore au bout de 2 semaines de traitement, l'encorafenib doit être interrompu jusqu'à une amélioration à un grade 0 ou 1, puis repris à la même dose. |
| Grade 3 | L'encorafenib doit être interrompu jusqu'à une amélioration à un grade 0 ou 1 et repris à la même dose s'il s'agit de la première survenue ou alors repris à une dose réduite s'il s'agit d'une récidive de grade 3. |
| Grade 4 | L'encorafenib doit être définitivement arrêté. |
| Érythrodysesthésie palmo-plantaire | |
| Grade 2 | L'encorafenib doit être maintenu et des traitements d'appoint tels que des traitements locaux doivent être instaurés En l'absence d'amélioration malgré ces mesures dans les 2 semaines, l'encorafenib doit être interrompu jusqu'au retour à un grade 0 ou 1 puis repris à la même dose ou à une dose réduite. |
| Grade 3 | L'encorafenib doit être interrompu et des traitements d'appoint tels que des traitements locaux doivent être instaurés et l'état du patient doit être réévalué toutes les semaines L'encorafenib doit être repris soit à la même dose ou soit à une dose réduite une fois revenu à un grade 0 ou 1. |
| Uvéite comprenant iritis et iridocyclite | |
| Grade 1 à 3 | En cas d'uvéite de grade 1 ou 2 n'ayant pas répondu à un traitement oculaire spécifique (tel qu'un traitement local) ou en cas d'uvéite de grade 3, l'encorafenib doit être interrompu et un contrôle ophtalmologique doit être répété dans les 2 semaines. En cas d'uvéite de grade 1 qui s'améliore au grade 0, le traitement doit être repris à la même dose. En cas d'uvéite de grade 2 ou 3 qui s'améliore à un grade 0 ou 1, le traitement doit être repris à une dose réduite. |
| Sévérité des effets indésirablesa | Encorafenib |
| En l'absence d'amélioration après 6 semaines, le contrôle ophtalmologique doit être répété et l'encorafenib doit être définitivement arrêté. | |
| Grade 4 | L'encorafenib doit être définitivement arrêté et un suivi avec contrôle ophtalmologique doit être réalisé. |
| Allongement de l'intervalle QTc | |
| QTcF > 500 ms et allongement ≤ 60 ms par rapport à la valeur avant traitement | L'encorafenib doit être interrompu (consulter la rubrique Mises en garde et précautions d'emploi concernant la surveillance du QTc). L'encorafenib doit être repris à une dose réduite dès lors que l'intervalle QTcF ≤ 500 ms. L'encorafenib doit être définitivement arrêté au-delà d'une récidive. |
| QTcF > 500 ms et allongement > 60 ms par rapport à sa valeur avant traitement | L'encorafenib doit être définitivement arrêté (consulter la rubrique Mises en garde et précautions d'emploi concernant la surveillance du QTc). |
| Anomalies biologiques hépatique | |
| Aspartate aminotransférase (ASAT) ou alanine aminotransférase (ALAT) de grade 2 > 3x - ≤ 5x la limite supérieure de la normale (LSN) | L'encorafenib doit être maintenu. En l'absence d'amélioration dans les 4 semaines, l'encorafenib doit être interrompu jusqu'au retour à un grade 0 ou 1 ou aux valeurs initiales/avant traitement puis repris à la même dose. |
| Premier épisode de grade 3 (ASAT ou ALAT > 5x la limite supérieure de la normale et bilirubine plasmatique > 2x la LSN) | L'encorafenib doit être interrompu pendant 4 semaines au maximum. En cas de retour à un grade 0 ou 1 ou aux valeurs initiales, il doit être repris à une dose réduite. En l'absence d'amélioration, l'encorafenib doit être définitivement arrêté. |
| Premier épisode de grade 4 (ASAT ou ALAT > 20x la LSN) | L'encorafenib doit être interrompu pendant 4 semaines au maximum. En cas de retour à un grade 0 ou 1 ou aux valeurs initiales, il doit être repris à une dose réduite. En l'absence d'amélioration, l'encorafenib doit être définitivement arrêté. Ou alors l'encorafenib doit être définitivement arrêté. |
| Récidive de grade 3 (ASAT ou ALAT > 5x la limite supérieure de la normale ou bilirubine plasmatique > 2x la LSN) | L'arrêt définitif de l'encorafenib devra être envisagé. |
| Récidive de grade 4 (ASAT ou ALAT > 20x la LSN) | L'encorafenib doit être définitivement arrêté. |
a Critères communs de terminologie de l'institut national contre le cancer pour les événements indésirables (« National Cancer Institute Common Terminology Criteria for Adverse Events », NCI CTCAE) version 4.03
Tableau 3 : Adaptations posologiques recommandées pour l'encorafenib (utilisé en association au cetuximab) pour d'autres effets indésirables
| Sévérité des effets indésirables | Encorafenib |
| Effets indésirables de grade 2 récidivants ou mal tolérés Premier épisode d'effet indésirable de grade 3 | L'encorafenib doit être interrompu pendant 4 semaines au maximum. En cas de retour à un grade 0 ou 1 ou aux valeurs initiales, il doit être repris à une dose réduite. En l'absence d'amélioration, l'encorafenib doit être définitivement arrêté |
| Premier épisode d'effet indésirable de grade 4 | L'encorafenib doit être interrompu pendant 4 semaines au maximum. En cas de retour à un grade 0 ou 1 ou aux valeurs initiales, il doit être repris à une dose réduite. En l'absence d'amélioration, l'encorafenib doit être définitivement arrêté. Ou alors, l'encorafenib doit être définitivement arrêté. |
| Récidive d'effets indésirables de grade 3 | L'arrêt définitif de l'encorafenib devra être envisagé. |
| Récidive d'effets indésirables de grade 4 | L'encorafenib doit être définitivement arrêté. |
Durée du traitement
Il convient de continuer le traitement jusqu'à ce que le patient n'en tire plus de bénéfice ou en cas de survenue d'une toxicité inacceptable.
Omissions de doses
En cas d'oubli d'une dose d'encorafenib, la dose d'encorafenib ne doit pas être prise s'il reste moins de 12 heures avant la prise de la prochaine dose prévue.
Vomissement
En cas de vomissements après l'administration d'encorafenib, le patient ne doit pas prendre de dose supplémentaire et prendra la prochaine dose comme initialement prévu.
Populations spéciales Personnes âgées
Aucune adaptation posologique n'est nécessaire pour les patients âgés de 65 ans et plus (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
Les patients atteints d'une insuffisance hépatique légère à sévère peuvent être davantage exposés à l'encorafenib (voir rubrique Propriétés pharmacocinétiques).
L'administration d'encorafenib doit être effectuée avec précaution à une dose de 300 mg une fois par jour chez les patients atteints d'une insuffisance hépatique légère (classe A de Child-Pugh).
Aucune posologie ne peut être recommandée chez les patients atteints d'une insuffisance hépatique modérée (classe B de Child-Pughou sévère (classe C de Child-Pugh).
Insuffisance rénale
Aucun ajustement de dose n'est nécessaire chez les patients présentant une insuffisance rénale légère à modérée selon une analyse pharmacocinétique (PK) de population. Aucune donnée clinique n'est disponible avec l'encorafenib administré chez les patients présentant une insuffisance rénale sévère. Par conséquent, la nécessité éventuelle d'un ajustement de la dose ne peut pas être déterminée. L'encorafenib doit être utilisé avec précaution chez les patients présentant une insuffisance rénale sévère (voir les rubriques Mises en garde et précautions d'emploi et Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité de l'encorafenib n'ont pas encore été établies chez les enfants et les adolescents. Aucune donnée n'est disponible.
Mode d'administration
Braftovi s'administre par voie orale. Les gélules s'avalent entières avec de l'eau. Elles peuvent être prises pendant ou en dehors des repas. Il faut éviter l'administration concomitante d'encorafenib et de jus de pamplemousse (voir les rubriques Mises en garde et précautions d'emploi et Interactions avec d'autres médicaments et autres formes d'interactions).
Durée de conservation :
3 ans.
Précautions particulières de conservation :
À conserver à une température ne dépassant pas 30 °C.
À conserver dans l'emballage d'origine à l'abri de l'humidité.
Sans objet.
Symptômes
À des doses d'encorafenib comprises entre 600 et 800 mg une fois par jour, une insuffisance rénale (hypercréatinémie de grade 3) a été observée chez 3 patients sur 14. La dose la plus élevée a été administrée à la suite d'une erreur de dose chez un patient ayant reçu l'encorafenib à une dose de 600 mg deux fois par jour pendant 1 jour (dose totale de 1 200 mg). Les effets indésirables rapportés par ce patient étaient de grade 1 à type de nausées, vomissements et vision trouble tous résolutifs par la suite.
Conduite à tenir
Il n'y a pas de traitement spécifique en cas de surdosage.
Compte tenu de la liaison modérée de l'encorafenib aux protéines plasmatiques, l'hémodialyse serait probablement inefficace dans le traitement d'un surdosage par l'encorafenib. Il n'y a pas d'antidote connu contre l'encorafenib. En cas de surdosage, l'encorafenib doit être interrompu et la fonction rénale doit être surveillée ainsi que la survenue des effets indésirables. Un traitement symptomatique et des soins d'appoint doivent être instaurés si besoin.
Classe pharmacothérapeutique : agent antinéoplasique, inhibiteur de protéines kinases, code ATC : L01XE46
Mécanisme d'action
L'encorafenib est une petite molécule inhibitrice ATP-compétitive, puissante et hautement sélective de la kinase de RAF. La concentration inhibitrice 50 % (CI50) de l'encorafenib par rapport aux enzymes BRAFV600E, BRAF et CRAF était, respectivement de 0,35, 0,47 et 0,30 nM. La demi-vie de dissociation de l'encorafenib était > 30 heures et s'est traduite par une inhibition prolongée de pERK. Plus précisément, l'encorafenib inhibe la prolifération cellulaire in vitro et in vivo des cancers colorectaux exprimant la mutation V600E. L'encorafenib n'inhibe pas la signalisation RAF/MEK/ERK dans les cellules présentant un BRAF de type sauvage.
Association avec le cetuximab
L'activation du récepteur EGFR a été identifiée comme l'un des principaux mécanismes de résistance du CCR exprimant la mutation BRAF aux inhibiteurs du gène RAF. Une amélioration de l'efficacité antitumorale a été démontrée avec les associations d'un inhibiteur de BRAF et d'agents ciblant le récepteur EGFR, dans des modèles non cliniques. L'association d'encorafenib au cetuximab a augmenté l'inhibition de la croissance des cellules tumorales HT-29 (double mutation BRAFV600E et PIK3CAP449T) et SW1417 (simple mutation BRAFV600E).
Efficacité et sécurité clinique
Cancer colorectal métastatique exprimant la mutation BRAF V600E - Étude ARRAY-818-302
L'encorafenib en association au cetuximab a été évalué dans une étude multicentrique, randomisée, ouverte et contrôlée par traitement actif (ARRAY 818-302 BEACON CRC). Les patients admissibles
dans l'étude devaient être atteints d'un cancer colorectal métastatique exprimant la mutation BRAF V600E qui avait progressé après 1 ou 2 autres protocoles thérapeutiques. Les patients ne devaient pas avoir été déjà traités par des inhibiteurs RAF, des inhibiteurs MEK ou des inhibiteurs EGFR. La randomisation a été stratifiée selon l'indice de performance de l'Eastern Cooperative Oncology Group (ECOG), l'utilisation antérieure d'irinotécan et la source de cetuximab.
Au total 665 patients ont été randomisés (selon un rapport 1:1:1) pour recevoir l'encorafenib 300 mg administré par voie orale une fois par jour en association au cetuximab dosé selon son RCP approuvé (n = 220), ou l'encorafenib 300 mg administré par voie orale une fois par jour en association au binimetinib 45 mg administré par voie orale deux fois par jour et au cetuximab dosé selon son RCP approuvé (n = 224) ou un traitement de contrôle (irinotécan en association au cetuximab ou irinotécan/5- fluorouracil/acide folinique (FOLFIRI) en association au cetuximab, n = 221). Le traitement a été poursuivi jusqu'à la progression de la maladie ou une toxicité inacceptable.
Les critères d'évaluation en termes d'efficacité étaient la survie globale (SG) et le taux de réponse globale (TRG), tels qu'évalués par un comité indépendant de revue centralisée conduite en aveugle (BIRC), comparant l'encorafenib en association au cetuximab par rapport au traitement contrôle. Les autres mesures de l'efficacité sont résumées dans le Tableau 5 ci-dessous.
L'âge médian des patients était de 61 ans (de 26 à 91), 47 % étaient des hommes et 83 % étaient de race blanche. 51 % des patients présentaient un indice de performance ECOG de 0 à l'inclusion et 51 % avaient préalablement reçu de l'irinotécan. 46,8 % des patients avaient au moins 3 organes présentant des lésions tumorales à l'inclusion.
La durée médiane d'exposition était de 3,2 mois chez les patients recevant l'encorafenib en association au cetuximab et de 1,4 mois chez les patients traités par irinotécan/cetuximab ou FOLFIRI/cetuximab (bras contrôle). Chez les patients traités par l'association d'encorafenib et de cetuximab, la dose- intensité relative (RDI) médiane était de 98 % pour l'encorafenib et de 93,5% pour le cetuximab. Dans le bras contrôle, la RDI médiane était de 85,4 % pour le cetuximab, de 75,7 % pour l'irinotécan, et dans le sous-groupe de patients ayant reçu de l'acide folinique et du 5-fluorouracil, la RDI médiane était de 75,2 % et 75 %, respectivement.
Une amélioration statistiquement significative de la SG,du TRG et de la SSP a été démontrée avec l'encorafenib en association au cetuximab par rapport au traitement contrôle. Les résultats de l'efficacité sont résumés dans le Tableau 5 et les Figures 1 et 2.
Les résultats d'efficacité basés sur l'évaluation de l'investigateur étaient concordants avec l'évaluation centrale indépendante.
Tableau 5 : Étude ARRAY-818-302 : Résultats d'efficacité
| | Encorafenib en association au cetuximab | Encorafenib en association au binimetinib et au cetuximab | Irinotécan en association au cetuximab ou FOLFIRI en association au cetuximab (Contrôle) |
| Analyse du 11 février 2019 (analyse principale) | |||
| SG | |||
| Nombre de patientsa | 220 | 224 | 221 |
| Nombre d'événements (%) | 93 (42,3) | 90 (40,2) | 114 (51,6) |
| Médiane, en mois (IC à 95 %) | 8,4 (7,5 - 11,0) | 9,0 (8,0 ; 11,4) | 5,4 (4,8 ; 6,6) |
| HR (IC à 95 %)b (vs Contrôle) | 0,60 (0,45-0,79) | | |
| Valeur pb,c | | ||
| | 0,0002 | ||
| HR (IC à 95 %)b (vs Contrôle) | | 0,52 (0,37 ; 0,73) | |
| Valeur pb,c | | ||
| | < 0,0001 | ||
| Durée médiane du suivi, en mois (IC à 95 %) | 7,6 (6,4 ; 9,20) | 8,7 (6,8 ; 9,4) | 7,2 (6,1 ; 8,1) |
| | Encorafenib en association au cetuximab | Encorafenib en association au binimetinib et au cetuximab | Irinotécan en association au cetuximab ou FOLFIRI en association au cetuximab (Contrôle) |
| TRG (selon le BIRC) | |||
| Nombre de patientsd | 113 | 111 | 107 |
| TRG n (%) | 23 (20,4) | 29 (26,1) | 2 (1,9) |
| | (13,4 ; 29,0) | | |
| (IC à 95 %)e | | (18,2 ; 35,3) | (0,2 ; 6,6) |
| Valeur pb,c,f | < 0,0001 | | |
| Valeur pb,c,f | | < 0,0001 | |
| RC, n (%) | 6 (5,3) | 4 (3,6) | 0 |
| RP, n (%) | 17 (15,0) | 25 (22,5) | 2 (1,9) |
| MS, n (%) | 57 (50,4) | 41 (36,9) | 26 (24,3) |
| TCM, n (%) | 84 (74,3) | 76 (68,5) | 33 (30,8) |
| (IC à 95 %) | (65,3 ; 82,1) | (59,0 ; 77,0) | (22,3 ; 40,5) |
| SSP (selon le BIRC) | |||
| Nombre de patientsa | 220 | 224 | 221 |
| Nombre d'événements (%) | 133 (60,5) | 118 (52,7) | 128 (57,9) |
| SSP médiane, mois (IC à 95 %) | 4,2 (3,7 ; 5,4) | 4,3 (4,1 ; 5,2) | 1,5 (1,5 ; 1,7) |
| HR (IC à 95 %)b | 0,40 (0,31 ; 0,52) | | |
| Valeur pb,c | < 0,0001 | ||
| HR (IC à 95 %)b | | 0,38 (0,29 ; 0,49) | |
| Valeur pb,c | < 0,0001 | ||
| Analyse actualisée, analyse du 15 août 2019 | |||
| SG | |||
| Nombre de patientsa | 220 | 224 | 221 |
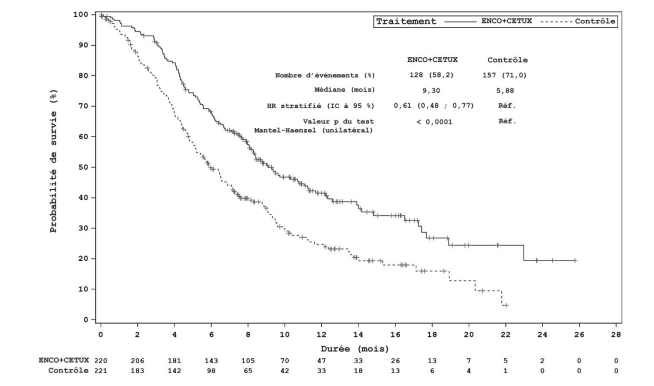
| Nombre d'événements (%) | 128 (58,2) | 137 (61,2) | 157 (71,0) |
| Médiane, en mois (IC à 95 %) | 9,3 (8,0 ; 11,3) | 9,3 (8,2 ; 10,8) | 5,9 (5,1 ; 7,1) |
| HR (IC à 95 %)b (vs Contrôle) | 0,61 (0,48 ; 0,77) | | |
| Valeur pb,c,g | | ||
| | < 0,0001 | ||
| HR (IC à 95 %)b (vs Contrôle) | | 0,60 (0,47 ; 0,75) | |
| Valeur pb,c,g | | ||
| | < 0,0001 | ||
| Durée médiane du suivi, en mois (IC à 95 %) | 12,3 (11,1 ; 14,1) | 14,1 (12,2 ; 16,1) | 12,9 (10,9 ; 14,6) |
| TRG (selon le BIRC) | |||
| Nombre de patientsa | 220 | 224 | 221 |
| TRG n (%) | 43 (19,5) | 60 (26,8) | 4 (1,8) |
| (IC à 95 %)e | (14,5 ; 25,4) | (21,1 ; 33,1) | (0,5 ; 4,6) |
| Valeur pb,c,f,g | < 0,0001 | < 0,0001 | |
| RC, n (%) | 7 (3,2) | 8 (3,6) | 0 |
| RP, n (%) | 36 (16,4) | 52 (23,2) | 4 (1,8) |
| MS, n (%) | 117 (53,2) | 98 (43,8) | 59 (26,7) |
| TCM, n (%) | 167 (75,9) | 168 (75,0) | 69 (31,2) |
| (IC à 95 %) | (69,7 ; 81,4) | (68,8 ; 80,5) | (25,2 ; 37,8) |
| | Encorafenib en association au cetuximab | Encorafenib en association au binimetinib et au cetuximab | Irinotécan en association au cetuximab ou FOLFIRI en association au cetuximab (Contrôle) |
| SSP (selon le BIRC) | |||
| Nombre de patientsa | 220 | 224 | 221 |
| Nombre d'événements (%) | 167 (75,9) | 157 (70,1) | 147 (66,5) |
| SSP médiane, en mois (IC à | 4,3 | 4,5 | 1,5 |
| 95 %) | (4,1 ; 5,5) | (4,2 ; 5,5) | (1,5 ; 1,9) |
| HR (IC à 95 %)b | 0,44 (0,35 ; 0,55) | | |
| Valeur pb,c, g | < 0,0001 | ||
| HR (IC à 95 %)b | | 0,42 (0,33 ; 0,53) | |
| Valeur pb,c, g | < 0,0001 | ||
IC = intervalle de confiance ; RC = réponse complète ; DdR = durée de la réponse ; HR = Hazard ratio (rapport des risques instantanés) ; NA = non atteint ; TRG = taux de réponse globale ; SG = survie globale ; RP = réponse partielle ; MS = maladie stable ; TCM : taux de contrôle de la maladie (Réponse Complète +Réponse Partielle +Maladie stable +Non-Réponse Complète/Non-Progression de la maladie ; Non-Réponse complète/Non-Progression de la maladie s'applique uniquement aux patients sans maladie non mesurable qui n'ont pas atteint la Réponse Complète ou n'ont pas une Progression de la maladie)
a Phase III randomisée, Jeu d'analyse complète
b Stratifié par IP ECOG, source du cetuximab et utilisation préalable d'irinotécan au moment de la randomisation
c Unilatéral
d Parmi les 331 premiers patients randomisés
e Méthode de Clopper-Pearson
f Test Cochran Mantel-Haenszel
g Valeur nominale de p
Figure 1 : Étude ARRAY-818-302 : Courbes de Kaplan-Meier de la survie globale (analyse du 11 février 2019)
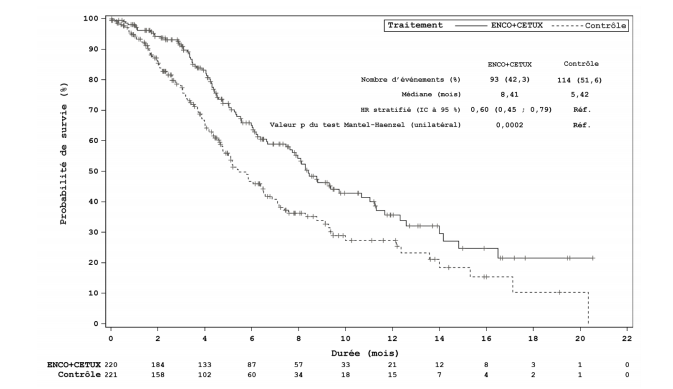
Figure 2: Étude ARRAY-818-302 : Courbes de Kaplan-Meier de la survie globale (analyse du 15 août 2019)
Qualité de vie (QdV) (analyse du 11 février 2019)
Le score FACT-C (Functional Assessment of Cancer Therapy-Colorectal cancer), le questionnaire de qualité de vie de l'Organisation européenne pour la recherche et le traitement du cancer (EORTC QLQ- C30), le questionnaire EQ-5D-5L (EuroQoL-5 Dimension-5 Level) et le questionnaire PGIC (Impression globale de changement du patient) ont été utilisés pour explorer les mesures rapportées par les patients, concernant la qualité de vie liée à la santé, les critères fonctionnels, les symptômes liés au cancer colorectal et les événements indésirables liés au traitement. Le temps médian jusqu'à détérioration définitive de 10 % du score de bien-être fonctionnel FACT-C était de 4,63 mois (IC à 95 % 3,94, 6,11) chez les patients traités par encorafenib en association au cetuximab et de 2,04 mois (IC à 95 % 1,87, 3,22) dans le bras contrôle (HR 0,57, IC à 95 % : 0,45, 0,72). Le temps médian jusqu'à détérioration définitive de 10 % du score d'état de santé global EORTC QLQ-C30 était de 4,6 mois (IC à 95% 3,81, 6,11) chez les patients traités par encorafenib en association au cetumixab et de 2,20 mois (IC à 95% 1,61, 3,06) dans le bras contrôle (HR 0,54, IC à 95% : 0,43, 0,69). Les analyses des échelles fonctionnelles émotionnelles et physiques du score EORTC QLQ-C30 ont donné des résultats cohérents avec les évaluations globales. Le score EQ-5D-5L était également favorable aux patients traités par encorafenib en association au cetuximab par rapport à ceux du bras de contrôle.
La proportion de patients ayant rapporté une amélioration du score PGIC aux cycles 2 et 4 était plus importante chez les patients traités par encorafenib en association au cetuximab que dans le bras contrôle.
Électrophysiologie cardiaque
Dans l'analyse de tolérance de l'étude en phase 3 (ARRAY-818-302) dans l'indication CCR, l'incidence d'un nouvel allongement de l'intervalle QTcF > 500 ms était de 3,2 % (7/216). Un allongement de l'intervalle QTcF > 60 ms comparé aux valeurs avant traitement a été observé chez 8,8 % des patients (19/216) dans le groupe encorafenib + cetuximab (voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi).
Population pédiatrique
L'Agence européenne des médicaments a accordé une dérogation à l'obligation de soumettre les résultats d'études avec encorafenib dans tous les sous-groupes de la population pédiatrique dans le cancer colorectal (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
La pharmacocinétique de l'encorafenib a été étudiée chez des sujets sains et chez des patients présentant des tumeurs solides, y compris des mélanomes cutanés avancés et inopérables ou métastatiques porteurs d'une mutation BRAF-V600E ou K, et chez des patients adultes atteints de CCR métastatique porteurs d'une mutation BRAF V600E. La pharmacocinétique de l'encorafenib s'est révélée approximativement linéaire par rapport à la dose après des doses uniques et multiples. Après une dose quotidienne unique répétée, l'état d'équilibre a été atteint dans un délai de 15 jours. Le facteur d'accumulation d'environ 0,5 est probablement dû à l'auto-induction du CYP3A4. La variabilité interindividuelle (CV en %) de l'ASC est comprise entre 12,3 % et 68,9 %.
Absorption
Après l'administration orale, l'encorafenib est rapidement absorbé avec un Tmax médian de 1,5 à 2 heures. Après une dose orale unique de 100 mg [14C] d'encorafenib chez des sujets sains, au moins 86 % de la dose d'encorafenib ont été absorbés. L'administration d'une dose unique de 100 mg d'encorafenib avec un repas riche en matières grasses et en calories a fait baisser la Cmax de 36 %, alors que l'ASC n'a pas changé. Une étude d'interaction médicamenteuse réalisée chez des sujets sains a indiqué que l'ampleur de l'exposition à l'encorafenib n'était pas altérée en présence d'un agent modifiant le pH gastrique (rabéprazole).
Distribution
L'encorafenib est modérément lié (86,1 %) aux protéines plasmatiques humaines in vitro. Après une dose orale unique de 100 mg [14C] d'encorafenib chez des sujets sains, le rapport de concentration sang-plasma moyen (ET) est de 0,58 (0,02) et le volume apparent de distribution (Vz/F) moyen (CV en %) de l'encorafenib est de 226 l (32,7 %).
Biotransformation
Après une dose orale unique de 100 mg [14C] d'encorafenib chez des sujets sains, le métabolisme s'est révélé la principale voie de la clairance pour l'encorafenib (environ 88 % de la dose radioactive récupérée). La principale réaction de biotransformation de l'encorafenib était la N-désalkylation. D'autres voies métaboliques majeures ont impliqué l'hydroxylation, l'hydrolyse de carbamates, la glycurono-conjugaison indirecte et la formation de conjugués au glucose.
Élimination
Après une dose orale unique de 100 mg [14C] d'encorafenib chez des sujets sains, la radioactivité a été éliminée équitablement à la fois dans les selles et dans l'urine (moyenne de 47,2 %). Dans l'urine, 1,8 %
de la radioactivité a été excrétée sous forme d'encorafenib. La clairance apparente (CL/F) moyenne (CV en %) d'encorafenib était de 27,9 l/h (9,15 %). La demi-vie terminale (T1/2) d'encorafenib médiane (min-max) était de 6,32 h (3,74 à 8,09 h).
Interactions médicamenteuses
Aucune interaction médicamenteuse n'a été mise en évidence entre l'encorafenib et le cetuximab.
Effet des enzymes CYP sur l'encorafenib
L'encorafenib est métabolisé par CYP3A4, CYP2C19 et CYP2D6. In vitro, il était attendu que CYP3A4 soit la principale enzyme contribuant à la clairance oxydative totale de l'encorafenib dans les microsomes hépatiques humains (~83,3 %), suivie par CYP2C19 et CYP2D6 (respectivement, ~16,0 % et 0,71 %).
Effet de l'encorafenib sur les substrats de CYP
Des expériences in vitro démontrent que l'encorafenib est un inhibiteur réversible relativement puissant des UGT1A1, CYP2B6, CYP2C9 et CYP3A4/5, ainsi qu'un inhibiteur temps-dépendant des CYP3A4. L'encorafenib a activé les CYP1A2, CYP2B6, CYP2C9 and CYP3A4 dans les cultures primaires d'hépatocytes humains. Des simulations d'administration concomitante de 450 mg d'encorafenib et de substrats de recherche pour CYP2B6, CYP1A2, CYP2C9, CYP2C19 and CYP2D6 le 1er et 15e jour ont toutes indiqué qu'aucune interaction pertinente du point de vue clinique n'est attendue. Concernant l'administration concomitante avec les substrats de CYP3A4 et UGT1A1 qui subissent une extraction intestinale, une interaction mineure à modérée est attendue.
Effet des transporteurs sur l'encorafenib
On a découvert que l'encorafenib était un substrat de la P-glycoprotéine (P-gp), un transporteur membranaire. Il est peu probable que l'inhibition de la P-gp engendre une augmentation importante d'un point de vue clinique des concentrations d'encorafenib puisque l'encorafenib présente une perméabilité intrinsèque élevée. Des études in vitro sur l'implication de plusieurs familles de transporteurs qui contribuent à l'absorption (OCT1, OATP1B1, OATP1B3 and OATPB1) ont été réalisées à l'aide d'inhibiteurs des transporteurs concernés. Les données indiquent que les transporteurs hépatiques contribuant à l'absorption ne sont pas impliqués dans la distribution de l'encorafenib dans les cultures primaires d'hépatocytes humains.
Effet de l'encorafenib sur les transporteurs
In vitro, l'encorafenib a inhibé le transporteur hépatique OCT1, mais il est peu probable qu'il soit un inhibiteur cliniquement efficace. Selon des études réalisées in vitro, il est possible que l'encorafenib inhibe les transporteurs rénaux OCT2, OAT1, OAT3 et les transporteurs hépatiques OATP1B1 et OATP1B3 à des concentrations cliniques. De plus, il se peut que l'encorafenib inhibe P-gp dans les intestins et le BCRP aux concentrations cliniques attendues.
Populations spéciales
Âge
Selon une analyse pharmacocinétique de population, il s'est avéré que l'âge était une covariable significative sur le volume de distribution de l'encorafenib, mais avec une grande variabilité. Étant donné la faible amplitude de ces changements et la grande variabilité, ces résultats ne sont probablement pas significatifs du point de vue clinique et aucun ajustement de dose n'est nécessaire chez les personnes âgées.
Sexe
Selon une analyse pharmacocinétique de population, le sexe n'apparaît pas comme étant une covariable importante sur la clairance ou le volume de distribution. Par conséquent, aucun changement majeur au niveau de l'exposition à l'encorafenib n'est attendu en fonction du sexe.
Poids corporel
Selon une analyse pharmacocinétique de population, il s'avère que le poids corporel est une covariable importante sur la clairance et le volume de distribution. Cependant, étant donné la faible amplitude du changement au niveau de la clairance et la grande variabilité du volume de distribution prédit dans le modèle, il est peu probable que le poids ait une influence considérable du point de vue clinique sur l'exposition de l'encorafenib.
Race
Il n'existe pas de différences cliniquement pertinentes de pharmacocinétique de l'encorafenib entre les patients de race asiatique et les patients de race non asiatique. Les données ne sont pas suffisantes pour évaluer les différences éventuelles au niveau de l'exposition de l'encorafenib dans d'autres races ou origines ethniques.
Insuffisance hépatique
Les résultats provenant d'un essai clinique spécifique indiquent des expositions totales de l'encorafenib 25 % plus élevées chez les patients atteints d'une insuffisance hépatique légère (classe A de Child- Pugh) comparés aux sujets dont la fonction hépatique est normale. Ceci se traduit par une augmentation de 55 % de l'exposition à l'encorafenib libre.
La pharmacocinétique de l'encorafenib n'a pas été évaluée sur le plan clinique chez les patients atteints d'une insuffisance hépatique modérée (classe B de Child-Pugh) à sévère (classe C de Child-Pugh). Comme l'encorafenib est principalement métabolisé et éliminé par le foie, selon le modèle pharmacocinétique physiologique, les patients atteints d'une insuffisance hépatique modérée à sévère peuvent être confrontés à des augmentations plus importantes de l'exposition que les patients atteints d'une insuffisance hépatique légère. Il n'est pas possible de recommander une dose chez les patients atteints d'insuffisance hépatique modérée ou sévère (voir rubriques Posologie et mode d'administration et Mises en garde et précautions d'emploi).
Insuffisance rénale
L'élimination rénale de l'encorafenib est minime. Aucune étude clinique officielle n'a été menée pour évaluer l'effet de l'insuffisance rénale sur la pharmacocinétique de l'encorafenib.
Dans une analyse pharmacocinétique de population, aucune tendance évidente au niveau de la CL/F de l'encorafenib n'a été observée chez les patients présentant une insuffisance rénale minime (eDFGe de 60 à 90 ml/min/1,73 m2) ou modérée (DFGe de 30 à 59 ml/min/1,73 m2) comparés aux sujets dont la fonction rénale est normale (DFGe ≥ 90 ml/min/1,73 m2). Une légère baisse de la CL/F (≤ 5 %) a été prédite chez les patients présentant une insuffisance rénale légère et modérée, ce qui n'est probablement pas significatif du point de vue clinique. La pharmacocinétique de l'encorafenib n'a pas été étudiée chez les patients présentant une insuffisance rénale sévère.
L'encorafenib a une influence mineure sur l'aptitude à conduire des véhicules ou à utiliser des machines. Des troubles de la vision ont été rapportés chez certains patients traités par l'encorafenib pendant les essais cliniques. Il faut recommander aux patients de ne pas conduire de véhicules ou d'utiliser de machines en cas de troubles de la vision ou de tout autre effet indésirable qui pourrait nuire à leur aptitude à conduire des véhicules ou à utiliser des machines (voir rubriques Mises en garde et précautions d'emploi et Effets indésirables).
Dans les études sur de toxicité chez le rat d'une durée de 4 et 13 semaines, des signes cliniques, une diminution du poids corporel, une diminution du poids des épididymes et de la prostate et des anomalies microscopiques au niveau des testicules, des épididymes, de l'estomac et de la peau ont été constatés. Une réversibilité partielle de ces anomalies a été observée après une période de récupération de 4 semaines. De plus, dans l'étude sur la toxicité chez le rat d'une durée de 13 semaines, des modifications pathologiques cliniques réversibles ont été constatées à des doses ≥ 100 mg/kg/j. Aucune dose sans effet nocif observable (NOAEL) n'a pu être établie pour l'étude de 4 semaines. La dose sans effet nocif observable (NOAEL) déterminée dans l'étude de 13 semaines était plus de 10 fois l'exposition à la dose thérapeutique chez l'Homme.
Dans l'étude sur la toxicité chez le singe d'une durée de 4 et 13 semaines, des épisodes isolés/sporadiques de vomissements et de diarrhée ainsi que des lésions ophtalmiques ont été observés à un niveau légèrement supérieur à l'exposition à la dose thérapeutique chez l'homme. Les lésions ophtalmiques étaient partiellement réversibles et consistaient en une séparation ou un décollement au niveau de la rétine entre la couche externe constituée par les bâtonnets et les cônes et l'épithélium pigmentaire de la rétine au niveau de la fovéa, la zone centrale de la macula. Cette observation était semblable à celle décrite chez les humains sous les termes de choriorétinopathie de type séreuse centrale ou rétinopathie séreuse centrale.
L'encorafenib ne s'est pas révélé génotoxique.
Aucune étude évaluant la fertilité n'a été réalisée avec l'encorafenib. Dans l'étude de toxicité d'une durée de 13 semaines, effectuée chez le rat, le traitement par encorafenib à une dose de 6 mg/kg/j niveau de dose équivalent à plus de 5 fois l'exposition humaine à la dose thérapeutique) a entraîné une diminution du poids des testicules et des épididymes, accompagnée d'une dégénérescence tubulaire et d'une oligospermie. Dans l'étude d'une durée de 13 semaines, une réversibilité partielle a été observée au niveau de dose le plus élevé (60 mg/kg/d).
L'étude sur le développement embryofoetal menée chez les rats a indiqué que l'encorafenib induisait une toxicité foetale avec des foetus de poids moins élevés et des retards de développement du squelette.
L'étude sur le développement embryofoetal menée chez les lapins a indiqué que l'encorafenib induisait une toxicité foetale avec des foetus de poids moins élevés et des changements temporaires au niveau du développement du squelette. Une dilatation de l'arc aortique a été observée chez certains foetus.
L'encorafenib s'est révélé phototoxique lors du test de phototoxicité in vitro 3T3 NRU (Neutral Red Uptake). L'encorafenib ne s'est pas révélé sensibilisant lors du test de sensibilisation in vivo réalisé chez la souris. L'ensemble de ces données indiquent que l'encorafenib présente un risque de potentiel phototoxique et un risque minime de sensibilisation aux doses thérapeutiques pour les patients.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I
Médicament à prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie
Médicament nécessitant une surveillance particulière pendant le traitement.
Médicament soumis à prescription hospitalière.
Gélule.
Coiffe de couleur chair opaque et corps blanc opaque, comportant un « A » stylisé, imprimé sur la coiffe et la mention « LGX 75mg » sur le corps. La gélule mesure environ 23 mm de long.
Polyamide/aluminium/PVC- plaquette en aluminium contenant 6 gélules.
Chaque boîte contient 42gélules.